Sindrome di Brugada o di morte improvvisa
La sindrome è stata descritta per la prima volta da autori italiani Andrea Nava, Bortolo Martini (1988).
E’ però conosciuta in tutto il mondo con riferimento al nome dei fratelli spagnoli Brugada, che descrissero nuovamente la sindrome nel 1992, cinque anni dopo gli articoli dei ricercatori italiani.
La sindrome di Brugada è una cardiopatia ad impronta aritmica causata dalla mutazione di geni che codificano per i canali ionici, i quali, essendo disfunzionanti, destabilizzano la normale attività elettrica del cuore.
Generalmente è una patologia su base ereditaria, anche se sono segnalate sporadiche mutazioni spontanee. Tale sindrome non dà sintomatologia ma può esordire tragicamente con morte cardiaca improvvisa, causata da aritmie ventricolari maligne ad insorgenza non prevedibile, solitamente in individui giovani e durante il sonno.
– Eziologia
La sindrome di Brugada è nella maggior parte dei casi a trasmissione autosomica dominante con una probabilità di trasmissione del 50% nel caso di un genitore affetto.
Ai dati attuali, sono almeno 40 i geni coinvolti, ma nel 15-30% dei test genetici la mutazione individuata è sul gene SCN5A localizzato sul cromosoma 3, che codifica per la subunità alfa del canale del sodio, il quale regola l’accoppiamento eccitazione-contrazione del cuore.
Quando risulta mutato, altera la corretta trasmissione dei segnali elettrici cardiaci generando possibili aritmie fatali..Epidemiologia
La prevalenza stimata della sindrome di Brugada nel mondo è di 5 casi su 10.000 individui, probabilmente sottostimata per difficoltà di diagnosi. L’incidenza è di circa 8 volte maggiore nel sesso maschile.
I decessi si verificano tra i 25 ed i 55 anni d’età. Tra tutti i soggetti con sindrome di Brugada, solo un numero esiguo va incontro a morte improvvisa.
– Sintomatologia
La patologia è nella maggior parte dei casi asintomatica.
Ciò che desta preoccupazione è l’imprevedibilità delle aritmie fatali con cui si può presentare, anche come prima manifestazione, in particolare torsioni di punta e conseguente degenerazione in fibrillazione ventricolare.
Altre possibili manifestazioni sono: sincope, extrasistoli sopraventricolari e ventricolari, fibrillazione atriale, blocchi seno-atriali o atrio-ventricolari.
– Diagnosi
La diagnosi è anamnestica e clinico-strumentale, basandosi soprattutto sulle anomalie riscontrate all’ECG, associate o meno alla sintomatologia (arresto cardiaco resuscitato, sincope, palpitazioni) e tramite indagine genetica in caso di familiarità.
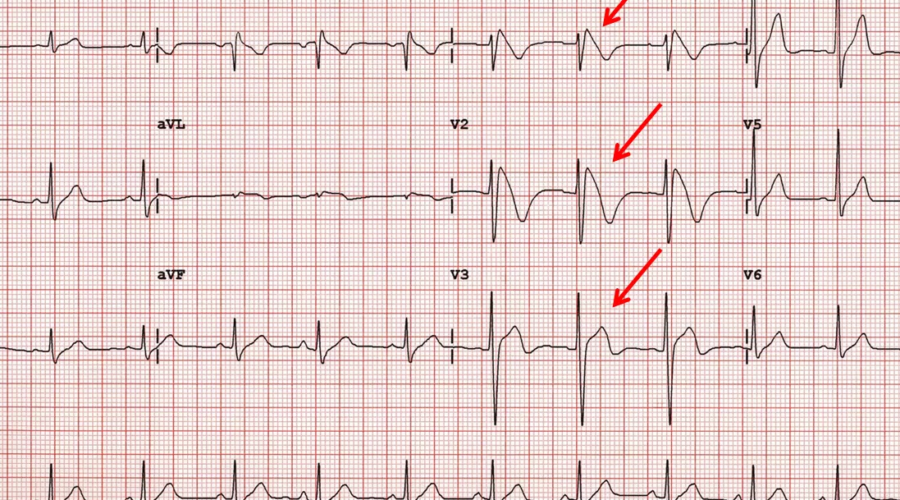
Le anomalie all’elettrocardiogramma vengono classificate in tipo 1, 2 o 3:
–Tipo 1 (coved type): con sopraslivellamento del punto J di almeno 2 mm ed una graduale discesa del segmento ST con morfologia a tenda ed un’onda T negativa in V1 e V2. Il tipo 1 (spontaneo o indotto) è l’unico ad essere specifico e patognomonico per Sindrome di Brugada.
–Tipo 2 (saddle-back pattern): con sopraslivellamento del punto J di almeno 2 mm ed un sopraslivellamento del tratto ST di almeno 1 mm, con un’onda T positiva o bifasica.
–Tipo 3 (saddle-back pattern): con un sopraslivellamento del punto J inferiore a 2 mm, un sopraslivellamento del tratto ST inferiore a 1 mm ed un’onda T positiva. Il tipo 3 è molto poco specifico considerando che risulta spesso presente anche in soggetti sani giovani e in atleti.
I tipi 2 e 3 sono anomalie elettrocardiografiche che pongono il sospetto per Sindrome di Brugada, per cui è raccomandata l’esecuzione in ambiente ospedaliero di un test provocativo alla Flecainide o Ajmalina per l’eventuale slatentizzazione del pattern di tipo 1.
Le manifestazioni ECG possono essere dinamiche o nascoste, o essere slatentizzate da stato febbrile (in tal caso è utile la somministrazione di paracetamolo in breve tempo per evitare l’aumento della temperatura corporea), ipo e iperkaliemia, ipercalcemia, assunzione di alcol o cocaina, o da determinati farmaci che bisogna assolutamente evitare (https://www.brugadadrugs.org/drug-lists/).
L’ esecuzione dell’ecocardiogramma è utile per l’esclusione di cardiopatie strutturali.
L’indagine molecolare è consigliata in caso di familiarità per sindrome di Brugada, anche se si rammenta che la patologia è connessa ad una mutazione genetica ereditabile nota soltanto nel 30-35% dei casi.
– Stratificazione del rischio
Per la stratificazione del rischio è fondamentale assumere un approccio multiparametrico che comprenda un’accurata raccolta clinico-anamnestica ed esami strumentali.
I soggetti ad alto rischio sono quelli con tipo 1 all’ECG basale e con 2 o più fattori di rischio (sincope, familiarità per morte improvvisa e studio elettrofisiologico positivo, BAV 1° grado), i rimanenti sono a basso rischio.
Considerando che i pazienti con sindrome di Brugada sono a rischio di morte improvvisa, lo studio elettrofisiologico (SEF) viene effettuato nei soggetti affetti, soprattutto in coloro che non hanno una specifica sintomatologia o familiarità per Brugada, allo scopo di riconoscere i soggetti a rischio di sviluppare spontaneamente aritmie e quindi candidabili all’impianto di defibrillatore cardiaco (ICD).
– Diagnosi differenziale
La diagnosi differenziale include una serie di patologie o condizioni che possono mimare la sindrome di Brugada, tra cui: blocco di branca destro atipico, cardiomiopatia aritmogena, ripolarizzazione preococe, pericardite acuta, ischemia ed infarto miocardico, pectus excavatum, embolia polmonare, iperkaliemia, ipercalcemia, distrofia muscolare di Duchenne.
– Trattamento
Il trattamento dipende dal livello di rischio del paziente.
· Pazienti a basso rischio aritmico: è necessario eseguire controlli cardiologici seriati, per verificare se emergono nuovi parametri di rischio aritmico. Utile è l’impianto di loop recorder, ossia un registratore sottocutaneo che monitora costantemente l’attività cardiaca, che permette di rilevare eventuali aritmie che potrebbero modificare la classe di rischio nel tempo ed il tipo di strategia terapeutica.
· Pazienti ad alto rischio aritmico o con arresto cardiaco resuscitato o con tachicardia ventricolare sostenuta
documentata: è fondamentale l’impianto di ICD, considerato come l’unico trattamento salvavita.
Alternative terapeutiche:
· Terapia farmacologica con Chinidina nei seguenti casi:
1) In coloro che rifiutano l’impianto di ICD, considerando che l’efficacia della Chinidina è inferiore all’ICD come protezione dalla morte cardiaca improvvisa.
2) Controindicazione all’impianto di ICD.
3) Bridge all’impianto di ICD.
4) Paziente con ICD e shock multipli (in caso di storm aritmico è indicato l’isoprotenerolo).
5) Trattamento di aritmie sopraventricolari.
· Ablazione epicardica transcatetere del substrato aritmico mediante radiofrequenze: in caso di pazienti ad alto rischio aritmico con impianto di ICD e ricorrenti episodi di sincope, tachicardia/fibrillazione ventricolare.